Identification of accurate bacteria is the most crucial step in microbiology, medicine, and environmental science. Conventional culture-based techniques are usually time-consuming and restricting, as most microbes fail to grow efficiently under laboratory conditions.
16S rRNA sequencing has revolutionized the process. By targeting the ribosomal RNA gene present in almost all bacteria, researchers can identify and classify microbes with much greater accuracy.
The true power of 16s rrna gene sequencing for bacterial identification comes from the fact that it is able to balance conserved regions (which are common among bacteria) with variable regions (which are specific to various groups).
This equilibrium enables scientists to pick up on a broad range of bacteria while continuing to differentiate between closely related strains.
we typically observe researchers and clinicians use this technique for accurate, culture-free insights into microbial populations.
What Is the 16S rRNA Gene?
For clarity on the process, it is important to learn about what is 16s sequencing and what 16S rRNA sequencing is based upon. The 16S ribosomal RNA (rRNA) gene has a length of approximately 1,500 base pairs and serves a structural function in the small subunit of the bacterial ribosome.
It is the conserved parts of this gene, however, that make it unique. These are maintained with great similarity in all bacteria, while the hypervariable regions (V1–V9) show great variation between species.
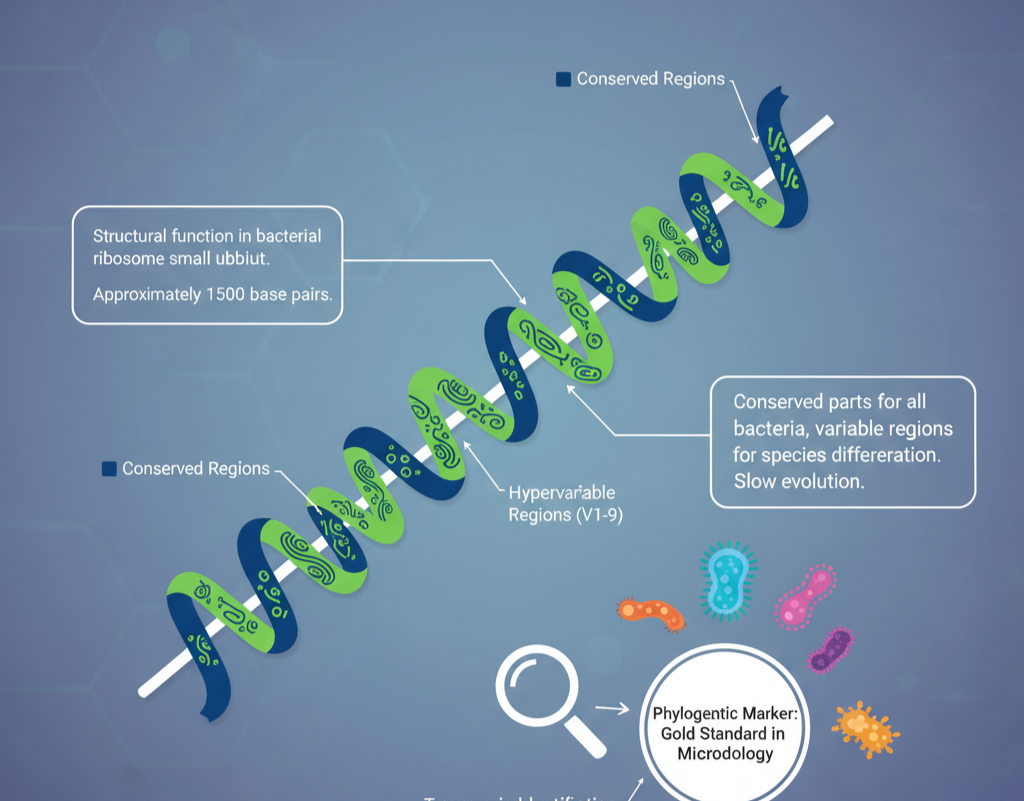
These variable regions function as molecular fingerprints, enabling scientists to identify taxonomic identity. Since it is found in all bacteria and shows slow evolution, the 16S rRNA gene sequencing has become the “gold standard” phylogenetic marker in microbiology.
How Does 16S Sequencing Identify Bacteria?
The basis of how 16S rRNA sequencing is done is simple but effective. The conserved areas of the 16S gene are amplified by the scientists using PCR (polymerase chain reaction) first, often through 16s rrna pcr for bacterial identification.
This makes the gene amplified from each bacterium in the sample. Second, the variable areas are sequenced and matched against curated reference databases.
This analysis enables taxonomic placement from high levels (such as phylum or genus) to species, as long as the genetic difference is high enough.
For most bacteria, this approach is correct to the genus level, but species-level decisions are occasionally hampered by similarities among close relatives.
Workflow Overview
The 16s rrna sequencing workflow typically takes a linear course:
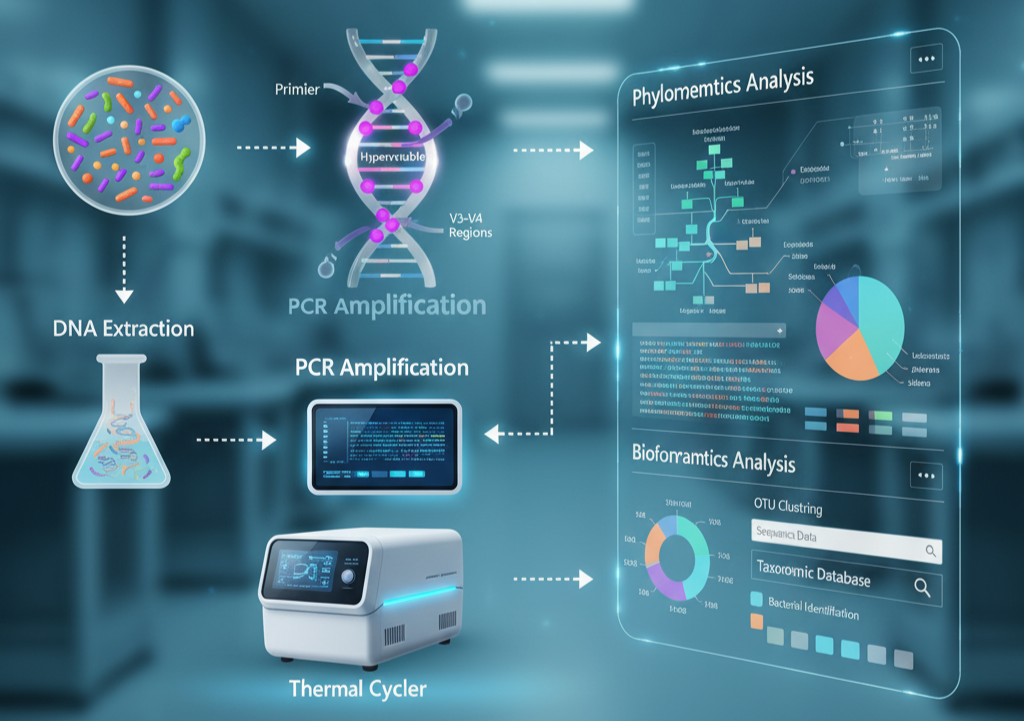
Sample Collection and DNA Extraction: DNA is extracted from bacterial cells, either pure cultures, patient, or environmental samples.
PCR Amplification: Target regions of the 16S gene are addressed by specific primers, such as hypervariable regions like V1–V3 or V3–V4.
Sequencing Methods:
- Single bacterial isolates are normally sequenced by Sanger sequencing.
- Next-Generation Sequencing (NGS) platforms are utilized for complicated microbial communities.

Bioinformatics Analysis: This involves quality control, the removal of sequencing artifacts or chimeras, clustering to ASVs (amplicon sequence variants) or OTUs (operational taxonomic units), and taxonomic classification using databases.
In Uncoded, a number of labs employ 16S rRNA sequencing kits that make this process easier by offering standardized reagents, primers, and analysis pipelines.
Selecting Variable Regions
Not all hypervariable regions are created equal. The selection is based on the research or diagnostic question. For instance, V3–V4 is a common region pair because it is a good balance between sequencing length and taxonomic resolution.
V1–V2 can be preferable for some bacterial clades, and V4, by itself, is ideal for high-throughput microbiome investigations.
Items like primer efficiency, compatibility with sequencing platforms, and supporting databases all play a role in determining the region to target. A badly selected region can generate unclear results, and thus careful selection is essential.
Databases and Taxonomic Classification
Sequencing data must be compared against reference databases for bacterial identification once they are produced. Resources that are widely used include SILVA, Greengenes, and RDP. These databases contain curated reference sequences to which researchers can compare their results.
For genus-level identification, a threshold of approximately 97% is usually used, and species-level calls might necessitate 99% or greater. Some methods also employ phylogenetic trees for more accurate placement when simple similarity scoring alone is not enough.
In Uncoded, professionals tend to emphasize open reporting, clearly declaring which database and threshold were utilized, as different databases can produce slightly variant identifications.
Strengths of 16S rRNA
The advantages of 16s rrna gene sequencing for bacterial identification are many:
- Culture Independence: It can detect bacteria that are unable to be cultured in the laboratory.
- Clinical Relevance: Quick pathogen identification in hospital environments, even from direct patient samples.
- Environmental and Microbiome Studies: Microbial community profiling in soil, water, gut, or skin with relative ease.

Limitations
Strong though it may be, the technique has significant caveats:
- Species-Level Ambiguity: Certain closely related bacteria, e.g., Escherichia coli and Shigella, have almost identical 16S sequences.
- Multi-Copy Bias: Most bacteria possess several copies of the 16S gene, which can skew abundance estimates.
- Lack of Functional Information: In contrast to shotgun metagenomics, this approach cannot uncover resistance genes, metabolic paths, or virulence factors.

Scientists employing Uncoded tools frequently couple 16S rRNA sequencing with supporting approaches to bridge these gaps.
When to Employ Alternatives or Complements
There are instances when other methods would be more appropriate. For instance:
- Whole-genome sequencing (WGS) yields full genetic information, with increased resolution and function data.
- Targeted gene panels can target selected resistance or virulence markers.
- Phenotypic tests might still be required for clinical laboratories for antibiotic susceptibility or for metabolic profiling.
The optimal strategy often combines 16S rRNA sequencing with culture, WGS, or clinical context to provide actionable results.
To wrap up
Strength of 16S rRNA sequencing for bacterial identification is in revealing microbial diversity that conventional techniques are unable to.
Through targeting a universally conserved but variable genetic marker, this technique has become a valuable instrument in microbiology, medicine, and environmental science.
At Uncoded, we observe researchers and clinicians increasingly using 16S rRNA sequencing kits and protocols to obtain faster, more precise, and culture-free bacterial identification. Though it has its downsides, particularly at the species level, it is one of the most affordable and commonly used tools in microbial research.
In the end, understanding how 16S rRNA sequencing works and when to use it in conjunction with other approaches enables scientists and clinicians to make informed, data-driven decisions.